> Towards the Atomic Scale Simulation of Intricate Acidic Aluminosilicate Catalysts
Céline Chizallet, IFPEN
Abstract
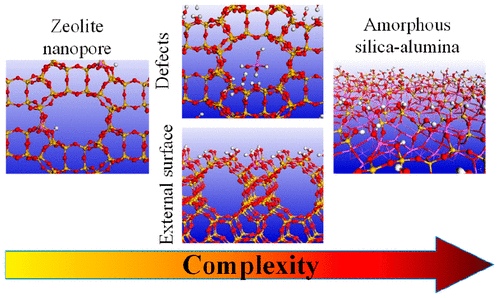
Zeolites are nanoporous aluminosilicates with well-defined crystalline structures, considered key assets in heterogeneous catalysis, with a broad range of industrial applications. Computational investigations dealing with zeolite catalysts have been undertaken for decades with simple models of the bulk sites, known to be bridging SiIV–OH–AlIV groups, in the case where the compensation cation is a proton. Real zeolite catalysts used in practice are however finite size and intricate objects, with external surface sites and defects, among other sources of complexity. Amorphous silica–alumina may also be obtained as a consequence of zeolite post-treatments or synthesized on purpose to obtain acid sites that are milder than those of zeolites. In the present Perspective, some of the achievements in the field of the atomic-scale simulation of intricate aluminosilicate catalysts (zeolites, amorphous silica–aluminas) of industrial relevance are reviewed. Emphasis is put on the simulation of the mechanisms of post-treatments of zeolites, and on the special structure and reactivity of acid sites at external surfaces of zeolites and on amorphous silica–alumina, that is shown to differ from the properties of the bulk bridging sites. Moreover, directions for future investigations are proposed.
ACS Catal. 2020, 10, 10, 5579–5601
Publication Date:April 14, 2020
https://doi.org/10.1021/acscatal.0c01136
Copyright © 2020 American Chemical Society