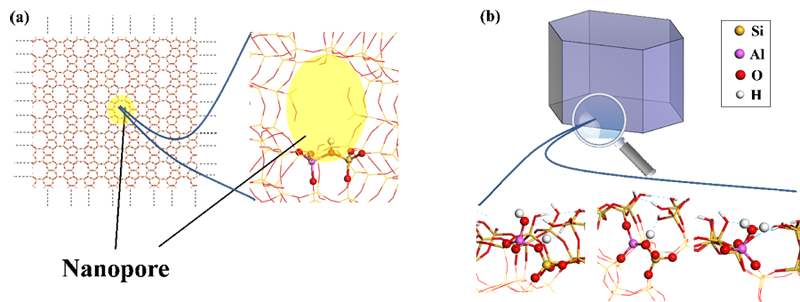
En dépit de leur importance, la structure et la réactivité des défauts à la surface externe des zéolithes réelles sont rarement traitées par le calcul quantique. Le modèle théorique du cristal parfait dont il se sert vise plutôt à connaître les nanopores des matériaux. Grâce à des calculs de modélisation quantique effectués sur la zéolithe ZSM-5, des travaux IFPEN ont pourtant démontré l’existence d’une variété de sites actifs bien plus grande à la surface que dans la nanoporosité interne.
De l’importance de connaître la surface externe des zéolithes
Les zéolithes sont des aluminosilicates cristallins nanoporeux servant dans de nombreuses applications actuelles et potentielles en catalyse, séparation et échange ionique. Dans le modèle du cristal parfait, ces solides présentent une structure bien définie. En cas d’emploi comme catalyseurs, connaître la structure nanoporeuse de ces matériaux, et l’effet de confinement associé, permet de comprendre et d’anticiper le comportement théorique des catalyseurs.
Toutefois, les zéolithes réelles, notamment celles employées à l’échelle industrielle, ne sont pas des objets parfaits et une grande partie de leurs propriétés découlent également de la présence de défauts. De ce point de vue, la taille finie des cristaux de zéolithes induit déjà une divergence par rapport au comportement théorique puisque la surface externe joue potentiellement un rôle important.
Ce dernier l’est d’autant plus lorsqu’une partie des réactifs à transformer (hydrocarbures longs, composés issus de la biomasse) sont trop encombrants pour accéder à la nanoporosité. Malgré cela, la structure et les sites actifs présents à la surface externe des zéolithes sont aujourd’hui méconnus.
Au-delà de la modélisation des nanopores : le nouvel éclairage du calcul quantique
Le calcul quantique est un outil puissant qui a fréquemment été mis en œuvre dans le cas des zéolithes modélisées comme des cristaux parfaits, appréhendés par la seule modélisation des nanopores (Figure a). Les travaux de calcul quantique traitant de la structure et de la réactivité des défauts, en particulier des surfaces externes des cristaux de zéolithes, sont beaucoup plus rares [1].
Des calculs récents de modélisation quantique (théorie de la fonctionnelle de la densité, DFT) apportent un éclairage nouveau sur la nature des sites de surface externe, leur acidité, et l’effet de confinement résiduel en surface externe (Figure b) [2,3].
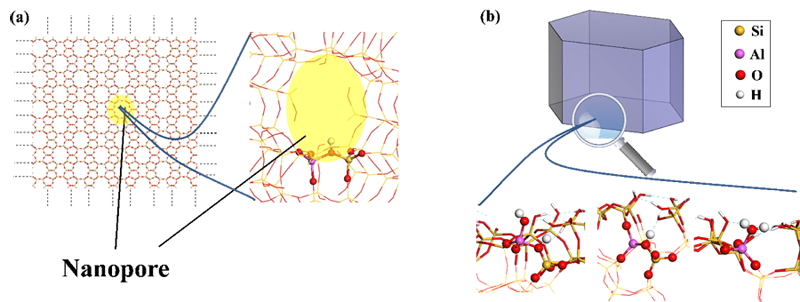
(b) Modélisation de la surface externe de la zéolithe ZSM-5, présentant une plus grande variété de sites acides de Brønsted et de Lewis au niveau des pores affleurant à la surface.
La zéolithe ZSM-5 à l’étude
Les derniers calculs de modélisation en date ont été menés dans le cadre d’un travail de thèse [4] qui a abordé le cas de la zéolithe ZSM-5, zéolithe parmi les plus étudiées et utilisées tant à l’échelle du laboratoire qu’à l’échelle industrielle. Les calculs effectués démontrent l’existence d’une variété de sites bien plus grande que dans la nanoporosité interne.
En effet, si cette dernière présente comme sites acides des OH dits pontés (Si-(OH)-Al, Figure a) de manière quasi exclusive, la surface externe offre en plus une variété d’hydroxyles dans des environnements divers (Si-OH, Al-OH) et de groupes Al-(H2O). Leur acidité de Brønsted (aptitude à céder des protons H+) est quantifiable grâce à la DFT, laquelle démontre l’existence à la fois de sites acides forts, moyens et faibles.
Les groupes Al-(H2O) donnent également lieu à des espèces AlIV et AlIII par déshydratation - elles apparaitront si la réaction que catalyse la zéolithe se produit à haute température-, qui sont des sites acides de Lewis, aptes à accepter des électrons de la part d’une espèce basique.
Enfin, un effet de confinement est toujours présent sur une partie de la surface externe, ce qui permet d’envisager un effet catalytique efficace même au niveau des pores ouverts
Des travaux complémentaires pour mieux comprendre la formation des défauts
Ces calculs quantiques ont été validés par un programme expérimental incluant d’une part des caractérisations spectroscopiques des sites de surface prédits et d’autre part l’étude des performances catalytiques, en utilisant des cristaux de tailles variables pour discriminer les effets respectifs de la surface interne et de la surface externe.
Des travaux complémentaires devraient être également réalisés dans le cadre du LCR CARMEN, afin de mieux comprendre la formation de défauts ponctuels et étendus (de type mésopores, de diamètre supérieur à 2 nanomètres), à partir des surfaces soit internes soit externes, dans la continuité de nos travaux précurseurs dans le domaine.
Références :
[1] Towards the Atomic Scale Simulation of Intricate Acidic Aluminosilicate Catalysts, C. Chizallet, ACS Catalysis, 10, 5579-5601, 2020. https ://doi.org/10.1021/acscatal.0c01136
[2] Ab initio simulation of the acid sites at the external surface of zeolite Beta, J. Rey, P. Raybaud, C. Chizallet, ChemCatChem, 9, 2176-2185, 2017. http://dx.doi.org/10.1002/cctc.201700080
[3] Environment, Stability and Acidity of External Surface Sites of Silicalite-1 and ZSM-5 Micro- and Nano-Slabs, -Sheets and –Crystals, L. Treps, A. Gomez, T. de Bruin, C. Chizallet, ACS Catalysis, 10, 3297−3312, 2020. https://doi.org/10.1021/acscatal.9b05103
[4] Thèse de Laureline Treps, 2017-2020, IFP Energies nouvelles, établissement d’inscription : Ecole Normale Supérieure de Lyon, sous la direction de C. Chizallet et le co-encadrement de T. de Bruin (IFPEN).
CONTACT :
Céline CHIZALLET
Chef de projet, calculs ab initio pour la Catalyse - IFPEN