Despite their importance, the structure and reactivity of defects on the external surface of real zeolites are rarely analyzed by quantum calculation. The theoretical model of the perfect crystal used in the latter is primarily aimed at gaining an understanding of the nanopores of materials. Thanks to quantum modeling calculations conducted on the ZSM-5 zeolite, IFPEN’s research has nevertheless demonstrated the existence of a much larger variety of active sites on the surface than in the internal nanoporosity.
The importance of knowing the external surface of zeolites
Zeolites are nanoporous crystalline aluminosilicates that can be used in numerous existing and potential applications in the fields of catalysis, separation and ionic exchange. In the perfect crystal model, these solids have a well-defined structure. When used as catalysts, knowing the nanoporous structure of these materials, along with the associated confinement effect, makes it possible to understand and anticipate the theoretical behavior of catalysts.
However, real zeolites, particularly those used on an industrial scale, are not perfect objects and many of their properties also stem from the presence of defects. From this point of view, the finite size of zeolite crystals already results in a divergence compared to the theoretical model since the external surface potentially plays an important role.
And this role is even more important when some of the reagents to be converted (long hydrocarbons, biomass-based compounds) are too bulky to access the nanoporosity. Despite this, the structure and the active sites present on the external surface of zeolites remain little understood.
Beyond nanopore modeling: the revelations of quantum calculation
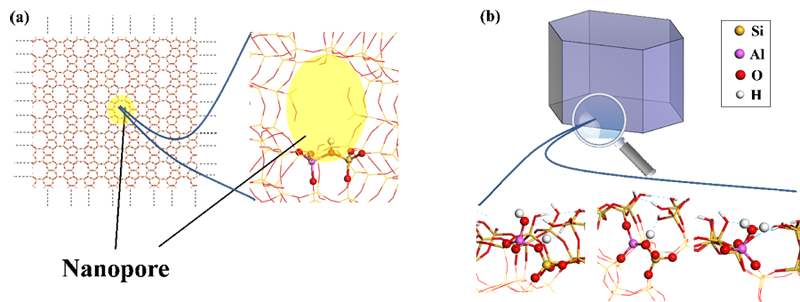
Quantum calculation is a powerful tool that has frequently been used for zeolites modeled as perfect crystals, based on nanopore modeling alone (Figure a). Quantum calculation research focusing on the structure and reactivity of defects, particularly on the external surface of zeolite crystals, is much scarcer [1].
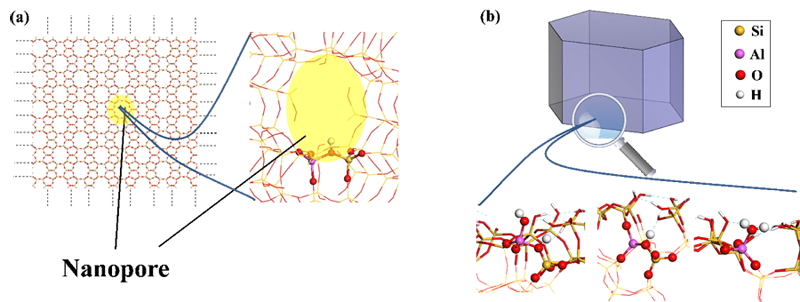
Recent quantum modeling calculations (density-functional theory, DFT) shed new light on the nature of the external surface sites, their acidity and the residual confinement effect on the external surface (Figure b) [2,3].
The ZSM-5 zeolite in the spotlight
The latest modeling calculations were conducted within the framework of a thesis [4] focusing on the case of the ZSM-5 zeolite, one of the most widely studied and used zeolites both in the laboratory and by industry. The calculations carried out demonstrate the existence of a much broader variety of sites than in the internal nanoporosity.
While the latter presents almost exclusively so-called bridged OH (Si-(OH)-Al, Figure a) acid sites, the external surface also offers a variety of hydroxyls in diverse environments (Si-OH, Al-OH) and Al-(H2O) groups. Their Brønsted acidity (capacity to release H+ protons) can be quantified using DFT, which demonstrates the existence of both strong, medium and weak acid sites.
Al-(H2O) groups also give rise to AlIV and AlIII species via dehydration - they will appear if the reaction catalyzed by the zeolite takes place at high temperature -, which are Lewis acid sites, with a capacity to accept electrons from a base species.
Finally, a confinement effect is still present on part of the external surface, making it possible to consider an effective catalytic effect even around open pores.
Complementary research for a better understanding of defect formation
Quantum calculations were validated using an experimental program incorporating, firstly, spectroscopic characterizations of predicted surface sites and, secondly, the study of catalytic performances, using crystals of variable sizes in order to differentiate between the respective internal and external surface effects.
Complementary research is also set to be conducted at the CARMEN joint research laboratory, in order to gain a better understanding of the formation of point and extended defects (mesopore-type, with a diameter of more than 2 nanometers), on external and internal surfaces, building on our pioneering research in the field.
References :
[1] Towards the Atomic Scale Simulation of Intricate Acidic Aluminosilicate Catalysts, C. Chizallet, ACS Catalysis, 10, 5579-5601, 2020. https ://doi.org/10.1021/acscatal.0c01136
[2] Ab initio simulation of the acid sites at the external surface of zeolite Beta, J. Rey, P. Raybaud, C. Chizallet, ChemCatChem, 9, 2176-2185, 2017. http://dx.doi.org/10.1002/cctc.201700080
[3] Environment, Stability and Acidity of External Surface Sites of Silicalite-1 and ZSM-5 Micro- and Nano-Slabs, -Sheets and –Crystals, L. Treps, A. Gomez, T. de Bruin, C. Chizallet, ACS Catalysis, 10, 3297−3312, 2020. https://doi.org/10.1021/acscatal.9b05103
[4] Thèse de Laureline Treps, 2017-2020, IFP Energies nouvelles, établissement d’inscription : Ecole Normale Supérieure de Lyon, sous la direction de C. Chizallet et le co-encadrement de T. de Bruin (IFPEN).
CONTACT :
Céline CHIZALLET
Project Leader, ab initio calculations for Catalysis - IFPEN